Tổng Quan Phòng Sạch Thiết Bị Y Tế
Phòng sạch thiết bị y tế là môi trường sản xuất được kiểm soát để đảm bảo thiết bị y tế không bị ô nhiễm hạt, vi sinh vật hoặc hóa chất — đặc biệt quan trọng với thiết bị cấy ghép (implant), dụng cụ xâm lấn và sản phẩm vô trùng. Ngành thiết bị y tế phải đồng thời đáp ứng cả kiểm soát hạt (như điện tử) và kiểm soát vi sinh (như dược phẩm).
Thị trường thiết bị y tế toàn cầu đạt ~$500 tỷ USD, Việt Nam đang trở thành điểm đến hấp dẫn cho sản xuất thiết bị y tế nhờ chi phí lao động cạnh tranh và chính sách ưu đãi FDI. Các tập đoàn như Medtronic, Terumo, Nipro đã đầu tư nhà máy với phòng sạch ISO 5-7 tại Việt Nam.
Phân Loại Thiết Bị Y Tế & Cấp Sạch Tương Ứng
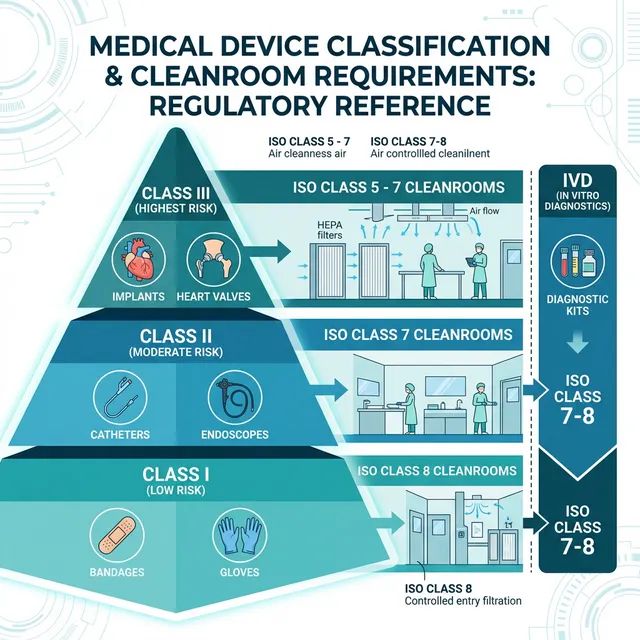
Phòng sạch sản xuất thiết bị y tế tuân thủ ISO 13485 và GMP. Yêu cầu cấp sạch phụ thuộc vào phân loại rủi ro của thiết bị:
| Phân loại | Mức rủi ro | Ví dụ | Cấp sạch | Vô trùng? |
|---|---|---|---|---|
| Class III | Cao nhất | Stent mạch, van tim nhân tạo, implant xương | ISO 5-7 | Bắt buộc |
| Class II | Trung bình | Ống nội soi, catheter, kim tiêm | ISO 7 | Đa số |
| Class I | Thấp | Băng gạc, găng tay, khẩu trang y tế | ISO 8 | Một số |
| IVD | Đặc biệt | Kit xét nghiệm, test nhanh, reagent | ISO 7-8 | Một số |
ISO 13485 — Yêu Cầu Phòng Sạch Chi Tiết
ISO 13485:2016 (Quality management systems for medical devices) là tiêu chuẩn quốc tế cho hệ thống quản lý chất lượng thiết bị y tế. Clause 6.4 (Work Environment) yêu cầu:
- Môi trường sản xuất phù hợp: Kiểm soát phù hợp với loại thiết bị — không quy định cấp ISO cụ thể nhưng phải chứng minh "phù hợp cho mục đích sử dụng"
- Documented requirements: Mọi yêu cầu kiểm soát môi trường phải được văn bản hóa
- Monitoring: Giám sát và ghi nhận điều kiện môi trường (hạt, vi sinh, nhiệt độ, ẩm)
- Cleaning and gowning: SOP vệ sinh phòng sạch và trang phục nhân viên
- Personnel hygiene: Đào tạo nhân viên về hành vi trong phòng sạch
💡 Lưu ý chuyên gia: Khác với GMP dược phẩm (quy định cấp A-B-C-D cụ thể), ISO 13485 để nhà sản xuất tự xác định cấp sạch phù hợp dựa trên risk assessment. Tuy nhiên, trong thực tế đánh giá bởi Notified Body (EU) hoặc FDA, nếu cấp sạch không đủ cho loại thiết bị, sẽ bị đánh nonconformance.
FDA 21 CFR 820 — Quality System Regulation
Nếu xuất khẩu thiết bị y tế sang Mỹ, nhà sản xuất phải tuân thủ FDA 21 CFR 820 (Quality System Regulation). Yêu cầu liên quan phòng sạch:
- §820.70(c) Environmental control: Môi trường sản xuất phải được kiểm soát đầy đủ để ngăn ô nhiễm sản phẩm
- §820.70(d) Personnel: Nhân viên phải mặc đồ bảo hộ phù hợp, tuân thủ quy trình vệ sinh
- §820.70(e) Contamination control: Phải có quy trình ngăn ô nhiễm sản phẩm bởi chất tẩy rửa, hóa chất, các sản phẩm khác
- Process validation: Quy trình sản xuất trong phòng sạch phải được validate — bao gồm cleaning validation và sterilization validation
EU MDR 2017/745
Quy định mới của EU (thay thế MDD 93/42/EEC) yêu cầu nghiêm ngặt hơn về technical documentation, post-market surveillance và clinical evaluation. Nhà sản xuất tại Việt Nam muốn xuất khẩu sang EU phải đạt CE marking theo EU MDR — phòng sạch là một phần quan trọng trong technical documentation.
Thiết Kế Phòng Sạch Thiết Bị Y Tế
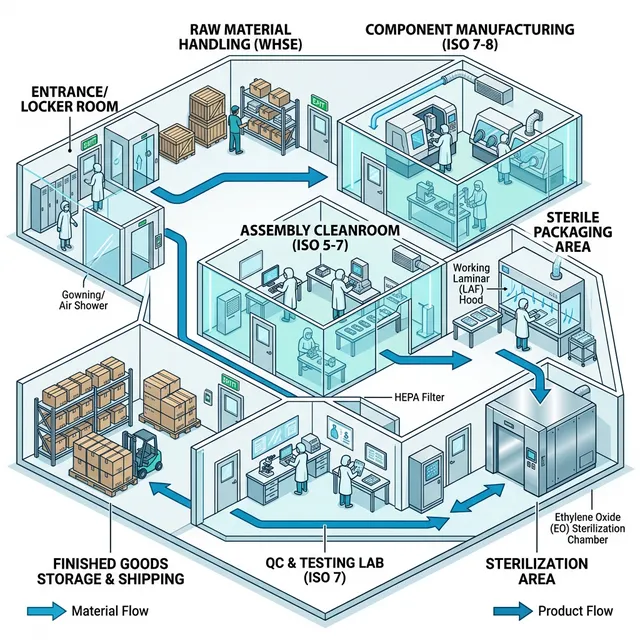
Đặc thù thiết kế so với các ngành khác
- Kiểm soát cả hạt VÀ vi sinh: Khác với điện tử (chỉ hạt) hay dược phẩm (chủ yếu vi sinh), thiết bị y tế yêu cầu kiểm soát đồng thời cả hai — đặc biệt với implant và dụng cụ xâm lấn
- Traceability: Khả năng truy xuất nguồn gốc từng lô sản phẩm. UDI (Unique Device Identification) là yêu cầu bắt buộc theo FDA và EU MDR
- Material compatibility: Phòng sạch không được gây ô nhiễm hóa chất lên thiết bị — vật liệu panel, sàn, sơn phải tương thích sinh học
- ESD control: Quan trọng cho thiết bị điện tử y tế (máy đo, monitor, thiết bị có vi xử lý)
Layout phòng sạch điển hình
- Khu vực gia công linh kiện: ISO 7-8 — cắt, mài, đúc, CNC machining
- Khu vực lắp ráp: ISO 5-7 — assembly trong LAF hoặc phòng sạch chuyên dụng
- Khu vực đóng gói vô trùng: ISO 5-7 — pouch sealing, blister packaging trong LAF
- Khu vực QC/QA: ISO 7-8 — kiểm tra ngoại quan, chức năng, vi sinh
- Khu vực tiệt trùng: EO chamber, gamma irradiation, steam autoclave
Đóng Gói & Tiệt Trùng (Sterilization)
Thiết bị y tế vô trùng phải được đóng gói trong bao bì barrier trước khi tiệt trùng. Quá trình đóng gói diễn ra trong phòng sạch ISO 7 trở lên:
Phương pháp tiệt trùng
| Phương pháp | Tiêu chuẩn | Ưu điểm | Phù hợp cho |
|---|---|---|---|
| EO (Ethylene Oxide) | ISO 11135 | Nhiệt độ thấp, xuyên thấu tốt | Nhựa, điện tử, catheter |
| Gamma irradiation | ISO 11137 | Xuyên thấu cao, không cần thông khí | Gạc, găng tay, syringe |
| Steam (autoclave) | ISO 17665 | Chi phí thấp, không hóa chất | Kim loại, thủy tinh |
| E-beam | ISO 11137 | Nhanh, không phóng xạ residual | Bao bì mỏng, thiết bị nhỏ |
| VHP | — | Nhiệt độ thấp, không residual | Surface sterilization |
Sterile barrier system
Bao bì vô trùng (sterile barrier system) theo ISO 11607 phải duy trì tính vô trùng của sản phẩm suốt thời hạn sử dụng. Yêu cầu: seal integrity test (burst test, dye penetration), accelerated aging test (40°C/75% RH), visual inspection. Đóng gói vô trùng phải thực hiện trong phòng sạch ≥ISO 7 với kiểm soát hạt và vi sinh.
Thẩm Định (Validation) Phòng Sạch Thiết Bị Y Tế
Khác với dược phẩm (IQ/OQ/PQ bắt buộc chi tiết), thiết bị y tế yêu cầu validation "phù hợp với rủi ro sản phẩm". Các loại validation quan trọng:
- Cleanroom qualification: Đo cấp sạch (theo ISO 14644-3), chênh áp, nhiệt độ, ẩm, ACH, recovery time
- Cleaning validation: Chứng minh quy trình vệ sinh phòng sạch và thiết bị đạt mức sạch yêu cầu
- Sterilization validation: Chứng minh phương pháp tiệt trùng đạt SAL 10⁻⁶ (thẩm định IQ/OQ/PQ)
- Packaging validation: Seal strength, burst test, accelerated aging
- Environmental monitoring: Chương trình giám sát hạt và vi sinh định kỳ
Xu Hướng Phòng Sạch Thiết Bị Y Tế Tại Việt Nam
Việt Nam đang thu hút mạnh FDI trong lĩnh vực thiết bị y tế:
- Medtronic: Mở rộng sản xuất tại Việt Nam — phòng sạch ISO 7 cho catheter và thiết bị tim mạch
- Terumo: Nhà máy sản xuất kim tiêm, syringe tại Bình Dương
- Nipro: Nhà máy dialyzer tại Long An — phòng sạch ISO 5-7
- Doanh nghiệp nội: MTG, Meditop — sản xuất khẩu trang, găng tay, kit xét nghiệm
Xu hướng chính: Chuyển từ sản xuất Class I (rủi ro thấp: găng tay, gạc) sang Class II (catheter, syringe) và IVD (kit xét nghiệm). Điều này đòi hỏi nâng cấp phòng sạch từ ISO 8 lên ISO 7 và đầu tư thêm sterilization facility.
💡 Lưu ý chuyên gia: EU MDR 2017/745 đã làm nghiêm ngặt hơn đáng kể yêu cầu cho thiết bị y tế xuất khẩu sang EU. Nhiều small-medium manufacturer mất CE marking vì không đủ nguồn lực. Tuy nhiên, đây cũng là cơ hội cho nhà sản xuất Việt Nam — nếu đầu tư phòng sạch và QMS đạt chuẩn, có thể trở thành OEM/ODM cho các thương hiệu EU.
Bạn cần tư vấn thiết kế phòng sạch sản xuất thiết bị y tế?
Đội ngũ chuyên gia của chúng tôi hỗ trợ từ ISO 13485, FDA compliance đến thiết kế phòng sạch và sterilization.
📞 Nhận tư vấn miễn phí ngay →